

Escherichia coli O157:H7 Recombinants
Abstract
There are 29 E. coli genomic sequences available, mostly related to studies of species diversity or mode of pathogenicity, including two genomes of the known clone O157:H7. However, genome studies of closely related clones intended to expose the details of evolutionary change have not been performed. Here we sequence the genome of a strain O55:H7, closely related to the major pathogenic clone Escherichia coli O157:H7 Recombinants, with published genomic sequences, and perform comparative genomic and proteomic analysis. We were able to assign most of the differences between the genomes to single mutations, recombination events, or lateral gene transfer events, in specific lineages.
Major differences include a type II secretion system present only on chromosome O55:H7, fewer type III secretion system effectors on O55:H7, and 19 phage genomes or phage-like elements on O55:H7 compared to 23 in O157:H7, with only three common to both. Many other changes were found in the O55:H7 and O157:H7 lineages, but overall there have been more changes in the O157:H7 lineages. For example, we found 50% more synonymous mutational substitutions in O157:H7 compared to O55:H7. The two strains also diverged at the proteomic level.
Synonymous mutational SNPs were used to estimate a divergence time of 400 years using a new clock rate, in contrast to 14,000 to 70,000 years using traditional clock rates. The same approaches were applied to three closely related extraintestinal pathogenic E. coli genomes, and similar levels of mutation and recombination were found. This study revealed for the first time the full range of events involved in the evolution of clone O157:H7 from its ancestor O55:H7 and suggested that O157:H7 arose recently. Our findings also suggest that E. coli has a much lower frequency of recombination relative to mutation than was observed in a comparative study of a Vibrio cholerae lineage.
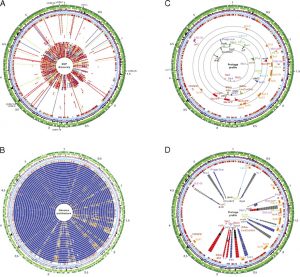
Results and Discussion
Phylogenomic analysis of the E. coli O157:H7 lineage.
Strain history and associated metadata for the 231 strains analyzed in this study are summarized in SI Materials and Methods and Dataset S1. To resolve the genetically homogeneous structure of the genome and study the genetic relatedness of E. coli O157:H7 strains, we implemented several strategies. The verotoxin profile of 231 strains of E. coli O157:H7, including the largest number of E. coli O157:H7, obtained from a single human outbreak (Data Set S1), revealed that the 191 strains associated with the outbreak of SP, three clinically derived strains that the Centers for Disease Control (CDC) considered outliers and not associated with the SP outbreak, and the eight strains obtained from the TB outbreak share characteristic Shiga toxin content stx1-, stx2+, stx2c+.
Notably, among the examined collection, seven unrelated human outbreak strains (EC1574, EC1582, EC1585, EC1588, EC1592, EC1610, EC4002, and EC508) assigned to clade 8 according to Manning et al. share this distinctive verotoxin pattern, but other genotypic and phenotypic data (Datasets S1 and S2) suggest that these clade 8 strains are not directly related phylogenetically to SP or TB outbreak strains.
Genotypic SNP profiles.
To validate our findings, we extended the genotypic profile and tested a total of 229 E. coli O157:H7 strains (Dataset S3). Strains underwent pyrosequencing-based screening of 19 canonical SNPs, which were chosen to differentiate the newly defined individual phylogenetic branches described above. The identified SNP panel allowed for genotypic clustering of closely related strains with high resolution and phylogenetic precision (Results and SI discussion).
However, SNP-based genotypic profiles are essential but not sufficient to establish genetic similarity. In this study, we have fully characterized the genetic states within group A and group B lineage I/II strains by cataloguing the genome architecture as well as the prevalence and location of prophages, revealing heterogeneity. genetics between these strains.